随机对照试验
【掌握】设计原则和基本类型
- 定义:随机对照试验是指根据研究目的,按照预先确定的研究方案将符合研究对象随机分配到试验组和对照组,进而分别接受相应的处理措施,并在一致条件下或同一环境中,同步进行研究、观测和比较组间效应,从而确定试验效果的一种试验性研究。
- 设计准则:
- 分配入组随机化。
- 组间具有可比性。
- 试验同步性和条件一致性。
- 试验实施一致性。
- 伦理学要求:
- 试验对象知情同意。
- 试验对象有权退出。
- 试验对象隐私保护。
【熟悉】随机对照试验的设计要点和研究中应注意的问题
- 设计要点:随机、对照、盲法。
- 样本量计算
- 影响样本量大小的主要因素
- I 型错误概率():出现假阳性错误的概率。 越小,样本量越大。通常将 设定为 0.05。
- 检验效能或把握度(): 为出现假阴性错误的概率(II 型错误概率), 为把握度。把握度越高,则所需样本量越大。通常将 设定为 0.90。
- 干预措施的预期效应大小:干预效应越大,所需样本量越小,反之亦然。
- 单侧检验或双侧检验:单侧检验比双侧检验所需样本量小。倘若凭借专业只是、前期研究有充分把握试验组优于对照组,用单侧检验,反之双侧检验。
- 资料类型:计数资料以治愈率、生存率、病死率等为分析指标时,基础发生率(p)越低,所需的样本量越大。而计量资料需要考虑总体均数和标准差等,标准差越大,所需样本量越大。
- 样本量的估计
- 计数资料的样本量估计:发病率、感染率、病死率、治愈率等。
- :对照组某结局的发生率;:试验组某结局的发生率;;:I 型错误概率的标准正态分布临界值;:II 型错误概率的标准正态分布临界值;n:每组所需样本量。
- 计量资料的样本量估计:均数、标准差等。
- :对照组均数;:试验组均数;:对照组标准差;n:每组所需样本量。
- 计数资料的样本量估计:发病率、感染率、病死率、治愈率等。
- 影响样本量大小的主要因素
- 资料整理:整理资料室需要特别注意以下情况
- 不合格的研究对象:一般要剔除,但提出不合格人数多于对照组,会造成结果误差。
- 不依从的研究对象:研究对象不遵试验要求。分为四种情况:I. 未完成 A 治疗或改为 B 治疗;II. 完成 A 治疗;III. 完成 B 治疗;IV. 完成 B 治疗但未完成 A 治疗。
- 处理方式:
- 意向治疗分析(ITT):比较 I + II 和 III + IV 组。这种反映原有试验意向的分析可能会低估其效果。即如果 ITT 分析表明 A 干预措施有效,则基本可以确认有效。
- 效力分析或完成治疗分析(PP):只比较 II 和 III,而不分析 I 和 IV。这种分析可能高估试验药物的生物效应。
- 接受治疗分析:比较 I + III 和 II + IV 组。由于研究对象未遵守试验要求、偏离或背离方案,组件缺乏可比性,造成结果可能失真。
- 处理方式:
- 失访。
- 注意事项
- 伦理问题
- 研究对象的选择问题:最好选择新发且发病频率较高的患者,这类患者对干预措施的效应反应明显,易于观察记录,且具有较好的反应性和敏感性。对孕妇、幼儿、老年人,一般不宜选为研究对象。
- 试验过程中的质量控制问题:对于研究对象不依从、失访等现象,对于研究对象的处理,需要一套严格的质量控制方案。
- 可行性问题:进行可行性分析,譬如预试验。
【熟悉】随机对照试验的优缺点
- 优点:
- 前瞻性的对照设计
- 组间可比性好
- 偏倚控制较好
- 病例诊断和干预措施标准化
- 局限性:
- 成本高
- 易失访
- 外部真实性受限
- 可能违背伦理
其他临床试验
【掌握】交叉试验
- 定义:试验中的试验组和对照组,在整个试验过程中通过前后两个阶段互相交叉的方式完成。也即分别先后接受两种不同试验措施的处理,最后评价试验结果的一种试验性研究。
- 应用范围:
- 适用于不易根治并需要药物长期维持的慢性疾病研究。如高血压、冠心病、心绞痛、支气管哮喘等。
- 在上市前新药临床试验中,为减少样本含量,I 期临床试验通常采用交叉试验来观察药物的毒副反应,以减少或消除个体间差异的影响。
- 设计模式和特点
- 交叉试验先将研究参与者随机分为两组,后进入两个试验阶段,第一阶段中,甲组-A,乙组-B。经过一段时间洗脱,甲组-B,乙组-A。最后比较两组的结果。
- 结果分析:对于定性资料采用配对 检验,对于定量资料采用配对 t 检验或符号秩和检验。
- 优缺点
- 优点:具有随机对照试验的优点,结果真实可靠。每个受试者经过两种方式处理,可以有自身前后比较,消除个体差异,也可获得组件比较结果。RCT 样本量计算公式适用于 COT,理论上可减少一半样本量。
- 缺点:应用范围受限。包括一定洗脱期。若试验周期长,容易发生失访、退出、依从性下降等。每个病例在第二阶段开始时很难保证病情处于试验第一阶段开始时的相似状态。
- 主要用途
- 用于研究药物应用先后顺序对治疗效果的影响。
- 用于研究药物的最佳配伍方案。
【掌握】自身前后对照试验
- 定义:每一个受试对象,先后接受试验和对照两种不同措施进行试验研究,最后将两次先后观测结果进行比较的一种设计方案。
- 适用范围:适用于慢性稳定或复发性疾病的临床治疗研究。
- 设计模式及特点:属于前瞻性研究设计。符合研究的纳入对象随机或非随机地在第一阶段接受一种措施的试验,然后经过一定的洗脱期后,研究参与者开始接受第二阶段的第二种措施。当完成试验后,将前后的试验结果进行分析比较。
- 结果分析
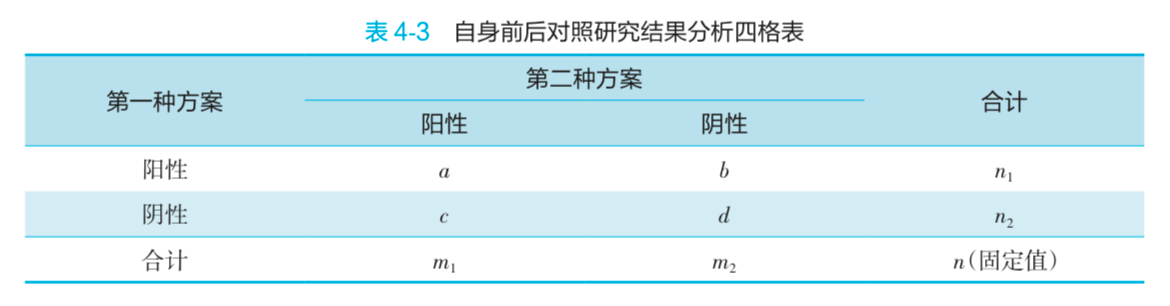
- 当 b + c >= 40 时,
- 当 b + c < 40 时,
- 优缺点
- 优点:研究参与者以自身为对照,可消除个体差异,可比性较好,节省样本量、时间和成本。每例参与者均有接受试验和对照两种措施的机会,具有公平性。减少了自愿者便宜和研究者意愿偏倚。可实现试验措施的标准化。试验中采用盲法并用随机方法安排前后干预措施,提高了结果可信度。
- 局限性:自身前后对照试验分为前后两个阶段,难以保证两阶段的起始点研究对象完全一致。试验范围有限。试验过程包括一定洗脱期。
【熟悉】其他类型临床对照试验
- 半随机对照试验:类似随机对照试验。唯一区别是研究对象按照半随机分配方式,即按照研究对象的生日、住院日、住院号等末位数字的奇偶数分为试验组合对照组。常常导致选择偏倚,且可靠性不如随机对照试验。因此在条件允许的情况下,尽量采用随机对照试验。
- 非随机同期对照试验:非随机分配入组。
- 历史性对照试验:将现有的干预措施的研究结果同过去的研究结果做非同期比较,主要用于评价现有方案或干预措施的作用。
- 单臂研究:仅有一个组的研究,未设置相应的对照。但可将他人或过去的研究结果与试验组比较。
伦理规范
【掌握】临床研究中的伦理原则
- 尊重原则:知情、自愿、保密。
- 不伤害原则:生理上的和心理上的。
- 有利原则:获益最大化,伤害最小化。研究得出确切结论应当确保每个患者都能够从中获益。
- 公平原则:分配公平、程序公平、回报公平。
【熟悉】伦理审查
- 文件:《人体生物医学研究国际道德指南》和《赫尔辛基宣言》。
- 伦理审查委员会:独立于研究者和赞助者的机构,负责对研究方案进行伦理审查。
- 伦理审查的内容
- 研究目的和设计
- 研究对象的选择
- 知情同意
- 风险和获益
- 隐私保护
- 数据管理和监测
- 研究者的资格和责任
- 伦理审查的程序
- 提交申请
- 伦理审查委员会审查
- 审查结果反馈
- 修改和重新提交(如有必要)
- 批准和实施
- 伦理审查的类型
- 全面审查:涉及高风险研究或复杂研究设计。
- 简化审查:涉及低风险研究或简单研究设计。
- 免审查:涉及不需要伦理审查的研究,如回顾性研究、匿名调查等。
- 伦理审查的时间:通常在研究开始前进行,审查周期一般为 2-4 周。
医学研究证据的检索与收集
【掌握】构建临床研究问题
- 使用 PICOS 原则构建临床研究问题
- P:Patient,患者。
- I:Intervention,干预。
- C:Control,对照。
- O:Outcome,临床结局。
- S:Study design,研究设计。
- 使用 PICOS 原则构建检索策略。